Pode ocorrer em indivíduos de todas as idades e qualquer etnia, sendo que a faixa etária predominante de surgimento da doença encontra-se entre os 40 e 50 anos de idade, sendo comum em idades mais avançadas. Do mesmo modo que acontece em outras doenças auto-imunes, costuma afetar muito mais as mulheres do que os homens (nove vezes mais).
A causa ou causas dessa doença não foram ainda esclarecidas, mas é provável que múltiplos fatores estejam envolvidos, como genéticos, viróticos, hormonais ou a associação destes.
Existem duas formas dessa síndrome, são elas:
- -Síndrome de Sjögren Primário: quando não há a presença de outra doença auto-imune concomitantemente.
- -Síndrome de Sjögren Secundário: quando outras doenças auto-imunes encontram-se associadas, como a artrite reumatóide, o lúpus eritematoso sistêmico, a polimiosite, a esclerodermia, entre outras.
As manifestações clínicas clássicas são nomeadas como síndrome seco (sicca síndrome), que diz respeito à secura da boca e olhos, conseqüente da redução de produção de saliva e lágrima. É comum a ocorrência de hipertrofia glandular (das parótidas).
O xeroftalmia, termo técnico usado na medicina para definir olhos secos, traduz-se por ardor, comichão, sensação de areia ou corpo estranho e fotofobia. Não ocorre apenas a redução da produção de lágrima, mas também uma alteração da qualidade desta. É comum o aparecimento de dilatação das glândulas lacrimais, infecções oculares, blefarite, podendo levar a infecções mais severas, como úlceras da córnea, queratites ou queratoconjuntivite.
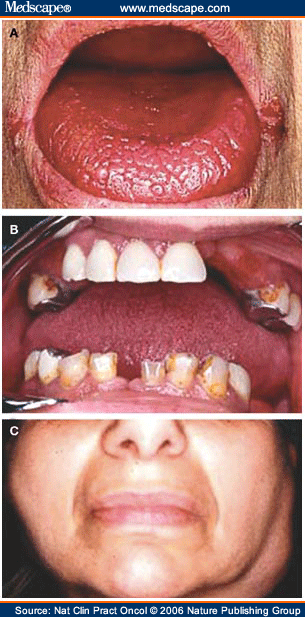
A xerostomia, termo técnico utilizado na medicina para descrever boca seca, apresenta como queixas a sensação de queimadura da boca, disfagia (dificuldade no ato de deglutir), especialmente para alimentos secos. Há uma maior tendência para o desenvolvimento de cáries, estomatite, mau hálito, anomalias do olfato e paladar. Aproximadamente 60% dos indivíduos acometidos pela síndrome apresentam hipertrofia das glândulas salivares.
Embora os alvos principais sejam as glândulas exócrinas, originando um processo inflamatório nestes, é muito freqüente a presença da secura de outros tecidos, como a pele, mucosa nasal e vaginal. Também pode causar problemas em outros locais do corpo, como músculos, articulações, pulmões, rins, tireóide, fígado, pâncreas, estômago, nervos e cérebro, resultando em diferentes sintomas relacionados aos órgãos envolvidos. Alterações psiquiátricas, como mudanças de humor, psicose, demência ou crises de pânico também podem estar presentes.
O diagnóstico é feito com base, principalmente, no quadro clínico, juntamente com os resultados obtidos de exames laboratoriais, de imagem e histológicos. O estudo imunológico também é importante, pois permite verificar se há a presença de anticorpos tipicamente encontrados nessa doença (anti-SSA e anti-SSB), embora não esteja necessariamente presente.
Não existe cura para esta síndrome e nem um tratamento específico para restabelecer permanentemente a função secretora das glândulas exócrinas. Deste modo, o tratamento é sintomático e de suporte, utilizando-se medicamentos, lágrimas artificiais, costicosteróides, entre outros. Podem ser utilizados anti-inflamatórios não-esteróides para tratar sintomas músculo-esqueléticos.
Recomenda-se molhar a boca com água freqüentemente, além de estimular a produção de saliva através de chicletes e rebuçados sem açúcar, pilocarpina em comprimidos, uso de bromexina ou saliva artificial em spray ou pastilhas. Para os lábios secos deve-se utilizar hidratantes labiais; para a secura da mucosa nasal, utilizar loções em cremes hidratantes; para a secura vaginal, utilizar cremes o gel lubrificantes.
Normalmente a evolução da síndrome de Sjögren é lenta e de forma benigna. No entanto, pode piorar ou se estabilizar, mas não ocorre remissão, como é possível em outras doenças auto-imunes.
Fontes:
http://pt.wikipedia.org/wiki/S%C3%ADndrome_de_Sj%C3%B6gren
http://www.drashirleydecampos.com.br/noticias/13304
http://www.abonet.com.br/abo/696/959-963.pdf
http://www.reumatorj.com.br/doencas/sjogren.pdf
http://www.spmi.pt/docs/nedai/sindromesjogren.pdf
http://pt.wikipedia.org/wiki/S%C3%ADndrome_de_Sj%C3%B6gren
http://www.drashirleydecampos.com.br/noticias/13304
http://www.abonet.com.br/abo/696/959-963.pdf
http://www.reumatorj.com.br/doencas/sjogren.pdf
http://www.spmi.pt/docs/nedai/sindromesjogren.pdf